
Investigadors de Fundació IMO han localitzat, per primera vegada, una mutació en el gen RP1 –fins ara lligat exclusivament a la retinosi pigmentària–, que provoca distròfia macular, un tipus de malaltia hereditària de la retina, que causa ceguesa a mig i llarg termini i que, de moment, no té tractament. Aquesta troballa s’ha publicat recentment a la revista British Journal of Ophthalmology.
Des del descobriment del gen RP1, fa 20 anys, només s'havia associat a retinosi pigmentària –una patologia retinal que afecta principalment les cèl·lules fotoreceptores conegudes com a bastons, de manera que provoca pèrdua de visió perifèrica, i que pot estar lligada a algun dels prop de 100 gens identificats fins ara com a possibles causants de la patologia.
30 gens associats a la distròfia macular
En el cas de distròfia macular, es coneixen al voltant de 30 gens capaços de desencadenar aquesta malaltia, que afecta un altre tipus de cèl·lules de la retina, els cons, i que comporta pèrdua de visió central. Precisament, la troballa dels investigadors de la Fundació IMO ha sorgit arran de la recerca de la mutació causant de diversos casos de distròfia macular, l'origen dels quals no es trobava en cap d'aquests 30 gens coneguts.
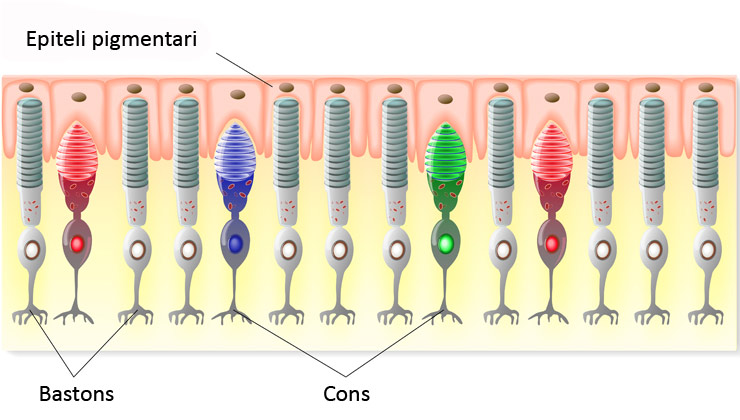
Las distròfies hereditàries de la retina n'afecten les cèl·lules fotoreceptores, ja siguin principalment els cons (com en el cas de la distròfia macular), els bastons (com passa a la retinosi pigmentària) o els dos tipus a la vegada.
Per trobar la mutació, es van examinar els 280 gens relacionats amb totes les distròfies de la retina (retinosi pigmentària, distròfia de cons i bastons, malaltia de Stargardt, amaurosi congènita de Leber, etc.). L'equip de la Fundació IMO va analitzar tots aquests gens en un centenar de famílies, entre elles 12 kuwaitians, i va localitzar, en aquesta dotzena, una variant patogènica comuna en el gen RP1, causant de la distròfia macular. Segons la Dra. Esther Pomares, responsable de l'equip que està darrere del descobriment, les 12 famílies de Kuwait "tenen un mateix haplotip (combinació d'al·lels) que s'hereta en bloc, conjuntament amb el gen RP1, on es produeix la mutació que hem descobert".
Avantpassat comú
Aquest fet posa de manifest que tots els afectats comparteixen un avantpassat comú, cap d'una tribu la descendència de la qual abasta a prop de 250.000 kuwaitians, cosa que ha sorprès el grup d'investigadors. "Es tracta d'un fenomen cultural molt allunyat del món occidental actual i que té un dels seus màxims exponents històrics en Gengis Kan, guerrer i conqueridor mongol del segle XIII, a qui s'atribueix un extens llinatge, després d'haver engendrat centenars de fills alguns dels quals, al seu torn, també van deixar una prolífica descendència", explica el Dr. Víctor Abad, de l'equip de la Fundació IMO.
Des del punt de vista científic, el descobriment permetrà ampliar el coneixement de les bases moleculars de la distròfia macular i obre la porta al futur tractament, amb teràpies gèniques o cel·lulars, dels pacients amb aquesta afectació l'origen genètic de la qual no s'havia trobat fins al moment. "Ara sabem que depenent de quina part del gen RP1 estigui alterada, també poden quedar afectats els cons, i no només els bastons, com es creia fins ara", afirma la Dra. Pomares.
Treball en equip
L'article sobre aquest avenç, publicat a la revista British Journal of Ophthalmology, el signen els membres del Departament de Genètica de l'IMO –Dra. Esther Pomares, Dr. Víctor Abad, Dra. Marina Riera, Sheila Ruiz i Pilar Méndez–, així com els oftalmòlegs, especialistes en retina de l’Institut, el Dr. Borja Corcóstegui i el Dr. Rafael Navarro.


